3.进行UTMOST分析
- 3.1 数据转换:将GWAS文件转换为适合UTMOST分析的格式
dat <- format_data_FinnGen(
GWASfile = "./1.data/finngen_R10_G6_MIGRAINE.gz", # 输入的GWAS文件路径
GWAS_name = "Migraine", # GWAS的名称
save_path = "./1.data", # 转换后数据的保存路径
type = "outcome", # 数据类型,表示这是结果数据
Twosample_dat = TRUE, # 是否保存Twosample格式数据
SMR_dat = TRUE, # 是否保存SMR格式数据
METAL_dat = TRUE # 是否保存METAL格式数据
)
- 3.2 单组织分析:进行单个组织的TWAS-UTMOST分析
data_utmost <- TWAS_UTMOST_single_tissue_test(
test_help = FALSE, # 是否显示帮助文档信息
GTEX_tissue = "all", # 使用所有组织数据
model_db_path = "./2.UTMOST/database_normalized_pruned", # 归一化后的模型数据库路径
covariance = "./2.UTMOST/covariance_GTEx8_normalized_pruned", # 归一化后的协方差矩阵路径
gwas_folder = "./1.data", # GWAS数据的文件夹路径
gwas_file_pattern = "Migraine_METAL_FinnGen.txt", # GWAS的文件名
snp_column = "SNP", # SNP列的名称
effect_allele_column = "effect_allele", # 效应等位基因列的名称
non_effect_allele_column = "other_allele", # 非效应等位基因列的名称
beta_column = "beta", # beta系数列的名称
pvalue_column = "pval", # p值列的名称
opt_arguments = NULL, # 额外的参数
save_path = "./2.UTMOST", # 保存结果的路径
cores = 14 # 运行使用电脑线程数
)
- 3.3 跨组织联合分析:进行跨组织的TWAS-UTMOST分析
#在运行此步之前,需要删除"./2.UTMOST"文件夹下的“all.csv"文件
data_GBJ <- TWAS_UTMOST_cross_tissue_test(
weight_db = "./2.UTMOST/database_normalized_pruned", # 权重数据的文件路径
cov_dir = "./3.UTMOST_GBJ/covariance_joint_GTEx8_normalized_pruned", # 协方差矩阵的文件路径
input_folder = "./2.UTMOST", # 输入文件夹路径
gene_info = "./2.UTMOST/gene_info.txt", # 基因信息文件路径
cores = 2, # 运行使用电脑线程数
output_dir = "./3.UTMOST_GBJ", # 输出目录路径
output_name = "Migraine" # 输出文件名
)
# 对UTMOST分析结果进行处理
dat_utmost_GBJ <- data_GBJ %>%
dplyr::left_join(data_info_GTEx_V8_GENE(), by = "ID") %>% # 将结果与GTEx基因信息数据合并
dplyr::mutate(FDR = p.adjust(p_value, method = "fdr")) %>% # 计算FDR值
dplyr::select(GENE, everything()) %>% # 对数据框的列进行重新排序
dplyr::arrange(FDR) # 按FDR值排序
# 将UTMOST分析结果保存为CSV文件
write.csv(dat_utmost_GBJ, file = "./3.UTMOST_GBJ/GBJ_res.csv", row.names = FALSE)
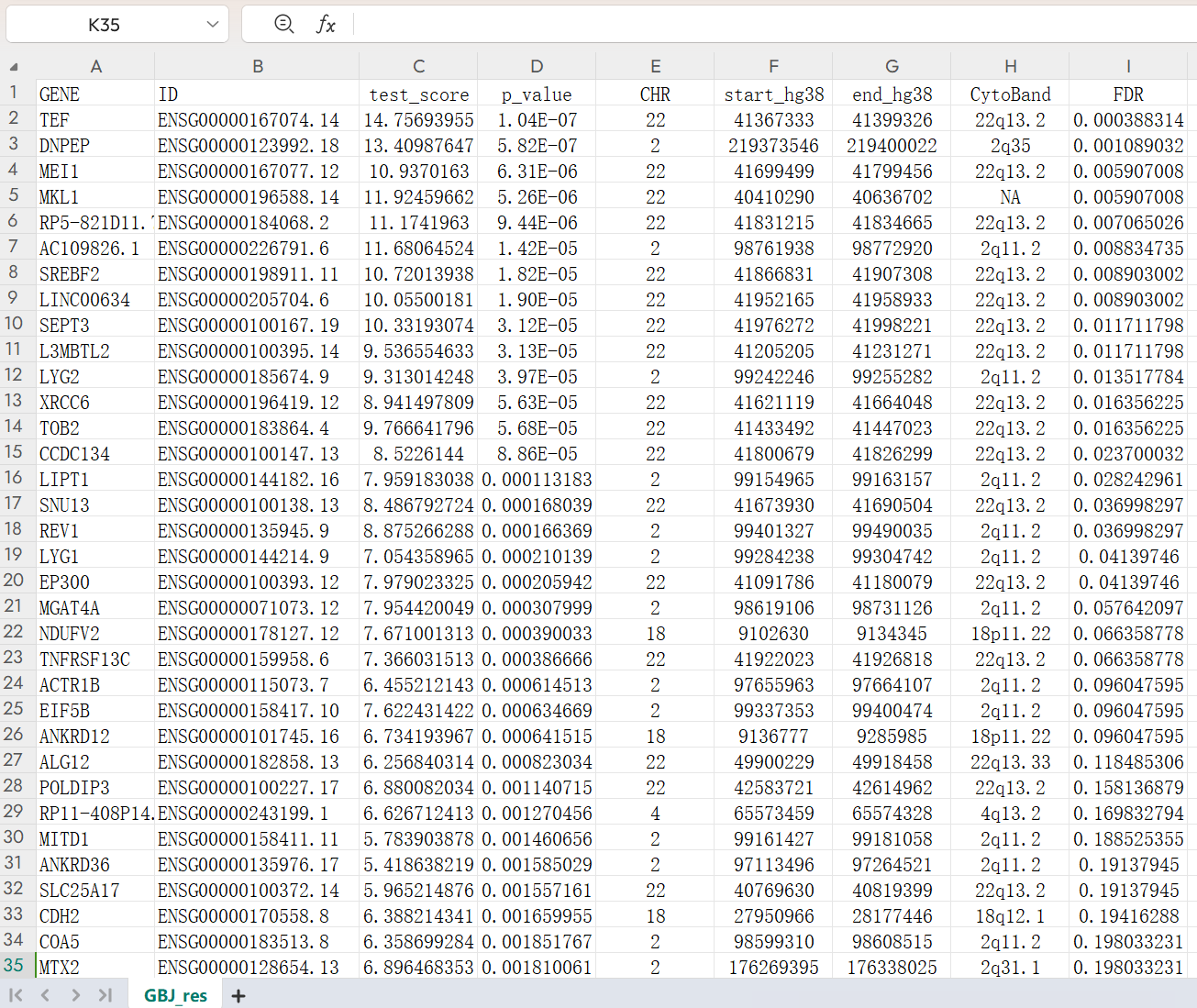
4.进行FUSION分析
- 4.1 数据转换:将GWAS数据转换为FUSION分析.sumstats.gz格式
TWAS_fusion_format_data(
GWASfile = "./1.data/Migraine_TwosampleMR_FinnGen.txt", # 输入的GWAS文件路径
N = NULL, # 样本量,如果为空则自动从数据中获取
save_name = "Migraine", # 保存的名称
save_path = "./1.data" # 保存路径
)
#此步骤耗时较长,约10余小时...
TWAS_fusion_Multi <- TWAS_fusion_Multi_test(
Sumstatsfile = "./1.data/Migraine.sumstats.gz", # 输入的结局sumstats.gz格式文件路径
weights_type = "nofilter.pos", # 权重数据类型
weights_dir = "./4.FUSION/WEIGHTS/", # 权重文件目录路径
resource = "GTEx_v8", # 资源数据库
ref_ld_chr = "./4.FUSION/LDREF/1000G.EUR.", # LD参考文件路径
start_chr = 1, # 起始染色体
end_chr = 22, # 结束染色体
coloc_pval = NULL, # 共定位p值(如果为空,则不使用共定位分析)
GWASN = NULL, # GWAS样本量(如果为空,则自动从数据中获取)
perm = 10000, # 置换检验迭代次数
PANELN = NA, # 面板样本量(如果为空,则自动从数据中获取)
opt_arguments = NULL, # 额外参数
remove_MHC = TRUE, # 是否去除MHC区域
FDR_method = "fdr", # FDR矫正方法
save_name = "Migraine", # 保存的名称
save_path = "./4.FUSION", # 保存路径
cores = 10 # 运行使用的电脑线程数
)
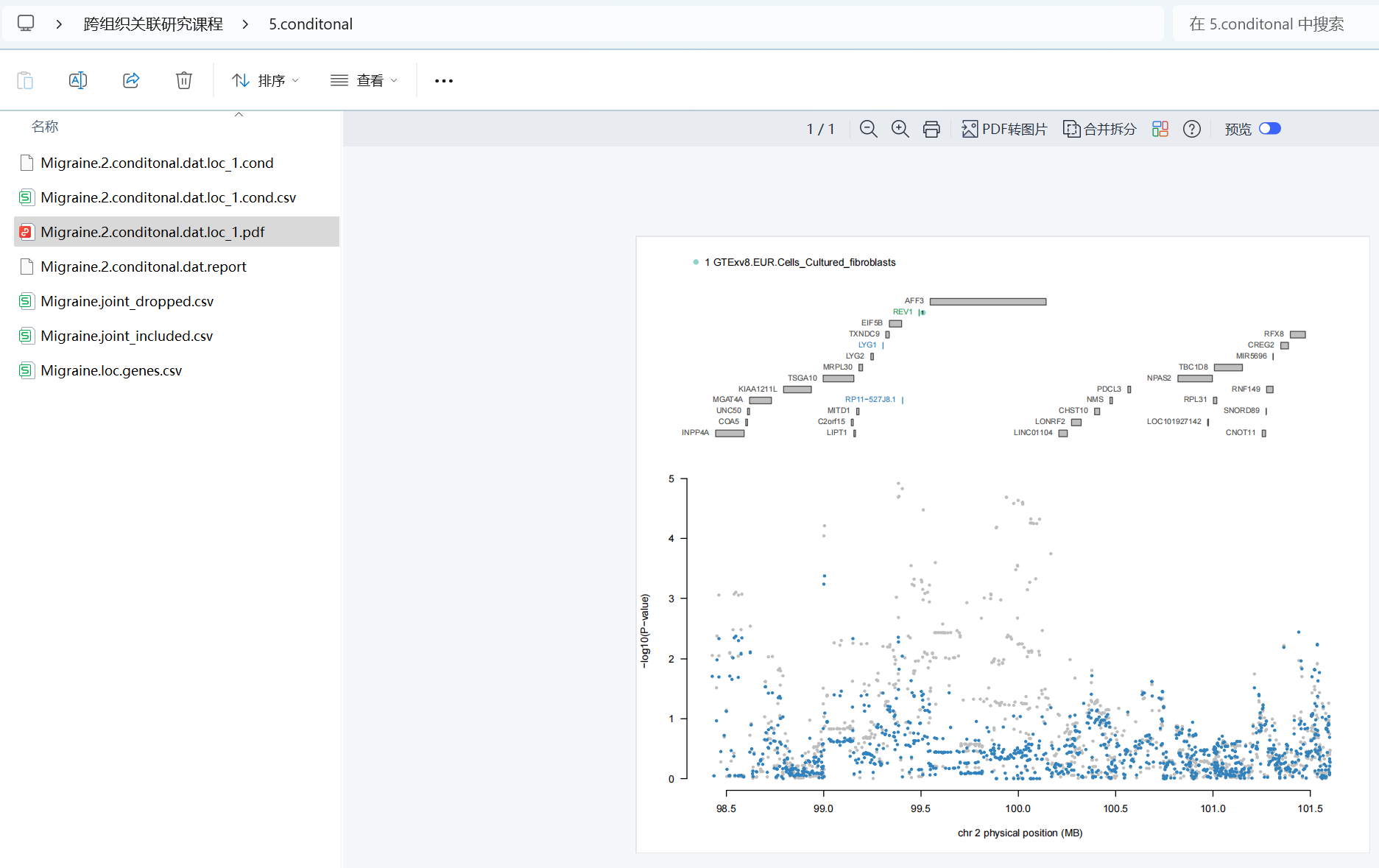
conditonal_test <- TWAS_fusion_conditonal_test(
test_help = FALSE, # 是否显示帮助文档信息
Sumstatsfile = "./1.data/Migraine.sumstats.gz", # 输入的结局sumstats.gz格式文件路径
inputfile = "./4.FUSION/GTExv8.EUR.Cells_Cultured_fibroblasts.nofilter.pos.sig.dat", # 显著性文件路径
ref_ld_chr = "./4.FUSION/LDREF/1000G.EUR.", # LD参考文件路径
start_chr = 1, # 起始染色体
end_chr = 22, # 结束染色体
locus_win_kb = 1000, # 基因组区域窗口大小(以kb为单位)
build = 38, # 基因组版本(38代表GRCh38)
plot_legend = "joint", # 图例类型
opt_arguments = NULL, # 额外参数
save_name = "Migraine", # 保存的名称
save_path = "./5.conditonal", # 保存路径
cores = 8 # 运行使用的电脑线程数
)
5.进行MAGMA分析
- 5.1 使用MAGMA软件进行基因层面和基因集合层面的关联分析
gene_based_dat <- MAGMA_gene_based(
GWAS_file = "./1.data/Migraine_METAL_FinnGen.txt", # 输入的GWAS文件路径
bfile_1000G = "./6.MAGMA/g1000_eur/g1000_eur", # 1000G参考数据路径
gene_loc = "./6.MAGMA/ENSGv110.coding.genes.txt", # 基因位置文件路径
set_annot = "./6.MAGMA/MSigDB_20231Hs_MAGMA.txt", # 基因集合注释文件路径
SNP_P_col = c(3, 10), # SNP p值列的位置
samplesize_col = "N", # 样本量列的名称
save_name = "Migraine", # 保存的名称
save_path = "./6.MAGMA" # 保存路径
)
Tissue_specific_dat <- MAGMA_Tissue_specific(
genes_raw = "./6.MAGMA/Migraine.genes.raw", # 原始基因数据路径
gene_covar = "./6.MAGMA/gtex_v8_ts_avg_log2TPM.txt", # 组织特异性表达数据路径
save_name = "Migraine", # 保存的名称
save_path = "./6.MAGMA/" # 保存路径
)
- 5.3 对MAGMA基因层面分析结果进行基因注释及曼哈顿图绘制
MAGMA_genes <- MAGMA_genes_Manhattanplot(
genes_out = "./6.MAGMA/Migraine.genes.out.txt", # 基因分析结果文件路径
gene_loc = "./6.MAGMA/ENSGv110.coding.genes.txt", # 基因位置文件路径
Manhtn_plot = TRUE, # 是否绘制曼哈顿图
threshold_sig = "bonferroni", # 显著性阈值调整方法
Manhtn_gene_sig = 10, # 曼哈顿图的基因标记标签的数目
signal_cex = 1, # 显著性基因图形圆点的大小
width = 9, # 图形宽度
height = 7, # 图形高度
save_name = "MAGMA_gsa" # 保存的名称
)
# 筛选FDR值小于0.05的基因
MAGMA_genes <- subset(MAGMA_genes, MAGMA_genes$P.fdr < 0.05)
# 对MAGMA通路富集分析结果图形可视化
plot_dat <- MAGMA_gsa_barplot(
gsa_out = "./6.MAGMA/Migraine.gsa.out.txt", # GSA结果文件路径
showNum = 50, # 显示的前50个通路
X_text_size = 5, # X轴文本大小
X_text_angle = 80, # X轴文本角度
Y_text_size = 10, # Y轴文本大小
save_plot = TRUE, # 是否保存图形
pdf_name = "gsa_plot", # PDF文件名
width = 9, # 图形宽度
height = 7, # 图形高度
save_path = "./6.MAGMA/" # 保存路径
)
# 对MAGMA组织特异性分析结果图形可视化
Tissue_specific_dat <- MAGMA_gsa_barplot(
gsa_out = "./6.MAGMA/Migraine.Tissue_specific.gsa.out.txt", # 组织特异性GSA结果文件路径
showNum = 54, # 显示的54个组织柱状图
X_text_size = 7, # X轴文本大小
X_text_angle = 70, # X轴文本角度
Y_text_size = 10, # Y轴文本大小
save_plot = TRUE, # 是否保存图形
pdf_name = "Tissue_specific_gsa_plot", # PDF文件名
width = 9, # 图形宽度
height = 7, # 图形高度
save_path = "./6.MAGMA/" # 保存路径
)
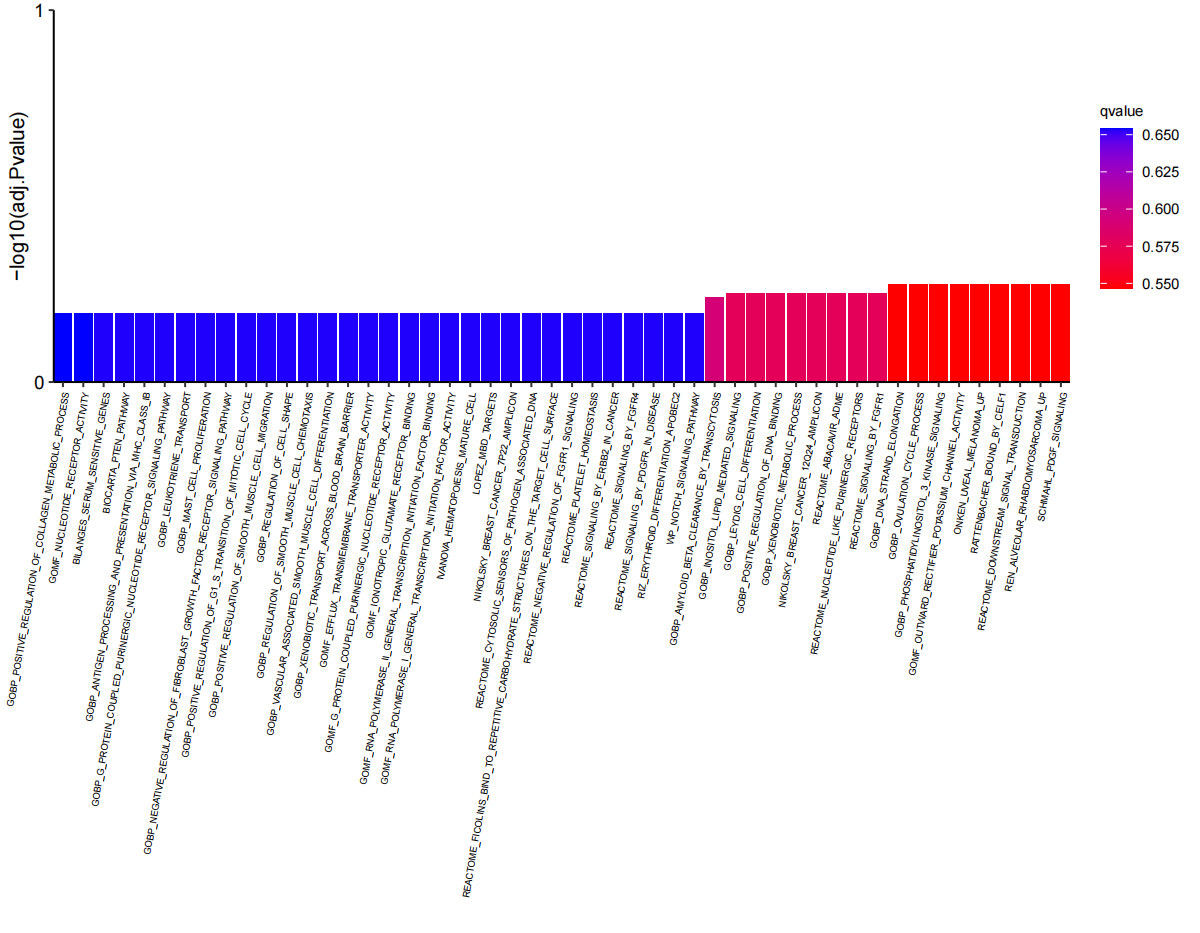
6.绘制韦恩图
- 对UTMOST、FUSION和MAGMA分析结果进行交集分析
venn_plot <- venn_plot(
files = c("./7.venn/UTMOST.txt", "./7.venn/FUSION.txt", "./7.venn/MAGMA.txt"), # 输入的结果文件路径
save_plot = TRUE, # 是否保存图形
save_name = "venn_plot", # 保存的名称
width = 9, # 图形宽度
height = 7, # 图形高度
save_path = "./7.venn" # 保存路径
)
7.孟德尔随机化(MR)分析
GTEx_V8_samplesize <- data_info_GTEx_V8_samplesize()
ciseQTL1 <- get_ciseQTL_GTEx_V8_Online(
Gene_name = "REV1", # 基因名称
tissues = "Whole Blood", # 组织类型
cis_wind_kb = 1000, # cis区域窗口大小(以kb为单位)
build = 38, # 基因组版本(38代表GRCh38)
save_path = "./8.MR" # 保存路径
)
ciseQTL1$gene <- "REV1(Whole Blood)" # 添加组织信息
ciseQTL2 <- get_ciseQTL_GTEx_V8_Online(
Gene_name = "REV1", # 基因名称
tissues = "Cells - Cultured fibroblasts", # 组织类型
cis_wind_kb = 1000, # cis区域窗口大小(以kb为单位)
build = 38, # 基因组版本(38代表GRCh38)
save_path = "./8.MR" # 保存路径
)
ciseQTL2$gene <- "REV1(Cells - Cultured fibroblasts)" # 添加组织信息
ciseQTL3 <- get_ciseQTL_GTEx_V8_Online(
Gene_name = "SREBF2", # 基因名称
tissues = "Testis", # 组织类型
cis_wind_kb = 1000, # cis区域窗口大小(以kb为单位)
build = 38, # 基因组版本(38代表GRCh38)
save_path = "./8.MR" # 保存路径
)
ciseQTL3$gene <- "SREBF2(Testis)" # 添加组织信息
# 合并所有的cis-eQTL数据
ciseQTL <- rbind(ciseQTL1, ciseQTL2, ciseQTL3)
# 按基因分割数据成list
ciseQTL <- split(ciseQTL, ciseQTL$gene)
# 保存合并后的cis-eQTL数据
save(ciseQTL, file = "./8.MR/ciseQTL.Rdata")
exp <- c()
for (i in names(ciseQTL)) {
dat <- format_dat(
ciseQTL[[i]], # 输入的cis-eQTL数据
type = "exposure", # 数据类型为暴露数据
phenotype_col = "gene", # 基因列的名称
snp_col = "SNP", # SNP列的名称
beta_col = "beta", # beta系数列的名称
se_col = "se", # 标准误列的名称
eaf_col = "eaf", # 等位基因频率列的名称
effect_allele_col = "effect_allele", # 效应等位基因列的名称
other_allele_col = "other_allele", # 非效应等位基因列的名称
pval_col = "pval", # p值列的名称
samplesize_col = "N", # 样本量列的名称
chr_col = "CHR", # 染色体列的名称
pos_col = "BP" # 位点列的名称
) %>%
clump_data_local_Online(
snp_col = "SNP", # SNP列的名称
pval_col = "pval.exposure", # p值列的名称
clump_method = "PVAL", # clump方法
beta_col = "beta.exposure", # beta系数列的名称
se_col = "se.exposure", # 标准误列的名称
clump_pval = 5e-08, # clump p值阈值
clump_kb = 10000, # clump窗口大小(以kb为单位)
clump_r2 = 0.001, # clump R^2阈值
pop = "EUR", # 参考人群为欧洲人群
bfile_1000G = "./1.data/1000G/EUR" # 1000G参考数据路径
)
exp <- rbind(exp, dat) # 合并所有格式化和clump后的数据
}
out <- modified_proxy_1000G(
snps = exp$SNP, # SNP列表
GWASfile = "./1.data/Migraine_TwosampleMR_FinnGen.txt", # GWAS文件路径
proxies = TRUE, # 是否寻找代理SNP
rsq = 0.8, # 代理SNP的R^2阈值
kb = 5000, # 代理SNP的窗口大小(以kb为单位)
nsnp = 5000, # 代理SNP的最大数量
maf_threshold = 0.3, # 最小等位基因频率阈值
bfile_1000G = "./1.data/1000G/EUR" # 1000G参考数据路径
)
- 7.5 对暴露和结果数据进行harmonisation(协调化)
dat <- TwoSampleMR::harmonise_data(exp, out)
# 进行孟德尔随机化(MR)分析
res <- xQTL_mr(dat, FDR_method = NULL, PVE = TRUE)
# 生成OR(比值比)结果
or <- TwoSampleMR::generate_odds_ratios(res)
xQTL_forest(
res, # MR分析结果
pvalsig = NULL, # 显著性p值阈值
ci_col = "#000000", # 置信区间颜色
ci_fill = "#000000", # 置信区间填充色
ci_lwd = 1, # 置信区间线宽
xlim = c(0.6, 1.2), # x轴范围
ticks_at = c(0.6, 0.7, 0.8, 0.9, 1, 1.1, 1.2), # x轴刻度
save_plot = TRUE, # 是否保存图形
plot_pdf = "forest_plot", # PDF文件名
width = 8, # 图形宽度
height = 5, # 图形高度
save_path = "./8.MR" # 保存路径
)
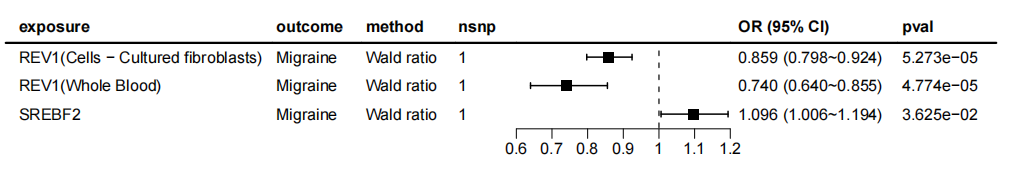
8.共定位分析
outcome_dat <- vroom::vroom("./1.data/Migraine_TwosampleMR_FinnGen.txt", show_col_types = FALSE)
# 加载之前保存的cis-eQTL数据
load("./8.MR/ciseQTL.Rdata")
coloc_summary <- c()
# 遍历每个基因进行共定位分析
for (j in names(ciseQTL)) {
dat_coloc <- coloc_abf(
exposure_dat = ciseQTL[[j]], # 暴露数据
outcome_dat = outcome_dat, # 结局数据
type_exposure = "quant", # 暴露类型为定量
col_pvalues_exposure = "pval", # 暴露数据中的p值列
col_N_exposure = "N", # 暴露数据中的样本量列
col_MAF_exposure = "MAF", # 暴露数据中的等位基因频率列
col_beta_exposure = "beta", # 暴露数据中的beta系数列
col_se_exposure = "se", # 暴露数据中的标准误列
col_snp_exposure = "SNP", # 暴露数据中的SNP列
col_chr_exposure = "CHR", # 暴露数据中的染色体列
col_pos_exposure = "BP", # 暴露数据中的位点列
prevalence_exposure = NA, # 暴露数据中的患病率
type_outcome = "cc", # 结局类型为病例对照
col_pvalues_outcome = "pval.outcome", # 结局数据中的p值列
col_N_outcome = "samplesize.outcome", # 结局数据中的样本量列
col_MAF_outcome = NA, # 结局数据中的等位基因频率列
col_beta_outcome = "beta.outcome", # 结局数据中的beta系数列
col_se_outcome = "se.outcome", # 结局数据中的标准误列
col_snp_outcome = "SNP", # 结局数据中的SNP列
prevalence_outcome = NA, # 结局数据中的患病率
save_stacked_dat = TRUE, # 是否保存绘制连锁区域图数据
build = 38, # 基因组版本(38代表GRCh38)
save_locus = FALSE, # 是否保存locus数据
title1 = paste0("eQTL ", j), # 图形标题1
title2 = "GWAS Migraine", # 图形标题2 请自行按照实际GWAS名字修改
width = 8, # 图形宽度
height = 5, # 图形高度
plot_pdf = "locuscompare", # PDF文件名
save_path = "./9.coloc" # 保存路径
)
# 提取和处理共定位分析结果
summary <- dat_coloc[["summary"]] %>%
dplyr::mutate(gene = j) %>%
dplyr::select(gene, everything())
# 绘制和保存共定位连锁区域结果图
dat_assoc <- coloc_stack_assoc_plot(
assoc = assoc,
bfile_1000G = "./1.data/1000G/EUR", # 1000G参考数据路径
SNP = SigSNP, # 显著SNP
build = 38, # 基因组版本(38代表GRCh38)
title1 = paste0("eQTL ", j), # 图形标题1
title2 = "GWAS Migraine", # 图形标题2
save_plot = TRUE, # 是否保存图形
width = 9, # 图形宽度
height = 10, # 图形高度
plot_pdf = paste0(j, "_plot"), # PDF文件名
save_path = "./9.coloc" # 保存路径
)
# 合并所有共定位分析结果
coloc_summary <- rbind(coloc_summary, summary)
}
print(coloc_summary)
write.csv(coloc_summary, file = "./9.coloc/coloc_res.csv", row.names = FALSE)
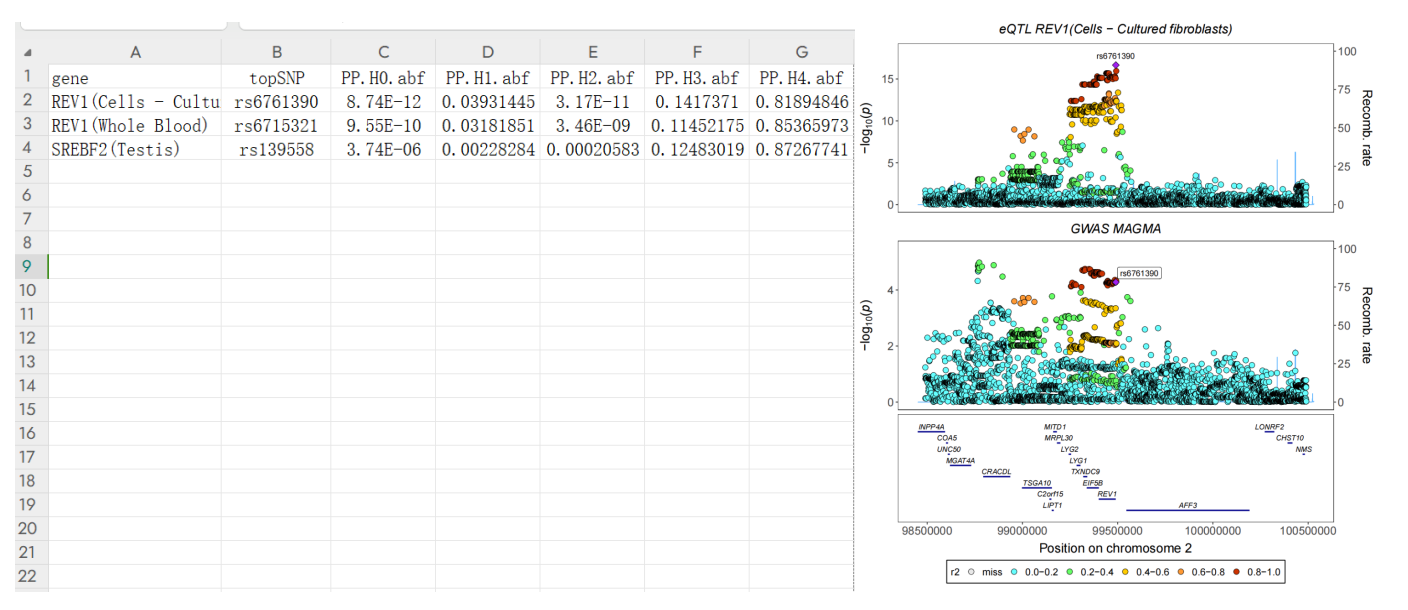
# 还可以联合SMR、FOCUS、TWAS其他算法、生信分析及湿实验等方法,丰富文章内容!
## 感谢各位小伙伴使用...